STAVER shows robust generalization across various large-scale DIA datasets¶
To further validate the reliability of STAVER’s results and underscore the robustness and inherent advantages of the STAVER algorithm, we applied STAVER to a much more diverse and larger-scale DIA dataset from the “ProCan-DepMap-Sanger project” (Gonçalves, et al. 2022, Cancer Cell). A total of 1,326 samples were included for analysis, including 84 samples of HEK293T cell lines used for quality control and 1,242 cancer cell samples derived from 9 typical cancer types (colorectal, SCLC, kidney, gastric, pancreatic, bladder, NSCLC, glioma, and hepatocellular cancer) (Figure RL2A-RL2B and Figure RL5A-RL5B).
We performed a comprehensive comparative analysis of the original data without the STAVER processing and the STAVER-processed data, with the main results focusing on - (I) the reproducibility and reliability of the STAVER-processed data, - (II) the robustness of the STAVER algorithm to uncover inherent biological differences, - (III) the reproducibility of the STAVER algorithm in identifying the previously reported tumor biomarkers, and - (IV) the robustness and broad applicability of the STAVER algorithm for disease diagnosis and classification.
[1]:
import pandas as pd
import numpy as np
import plotly.express as px
import plotly.io as pio
import plotly.graph_objects as go
import colorsys
import seaborn as sns
import matplotlib as mpl
import matplotlib.pyplot as plt
import scanpy as sc
import anndata
import os
import matplotlib.pyplot as plt
# python matplotlib export editable PDF
import matplotlib as mpl
mpl.rcParams['pdf.fonttype'] = 42
# mpl.rcParams['figure.dpi']= 150
import warnings
warnings.filterwarnings('ignore')
from plotly.offline import init_notebook_mode, iplot
init_notebook_mode(connected=True)
Metadata¶
[2]:
metadata = pd.read_excel("~/STAVER-revised/Pan-cancer-cell-lines/subset_949_samples.xlsx",index_col=0)
metadata
[2]:
| Automatic_MS_filename | Batch | Date | Instrument | Cell_line | SIDM | Tissue_type | Cancer_type | Cancer_subtype | Project_Identifier | |
|---|---|---|---|---|---|---|---|---|---|---|
| 519 | 191012_b36-t2-8_00di6_00jm7_m06_s_1 | P04 | 2019-10-12 | M06 | BFTC-905 | SIDM00989 | Bladder | Bladder Carcinoma | Urothelial carcinoma | SIDM00989;BFTC-905 |
| 521 | 191023_b61-t1-1_00dsu_00kp3_m03_s_1 | P04 | 2019-10-23 | M03 | BFTC-905 | SIDM00989 | Bladder | Bladder Carcinoma | Urothelial carcinoma | SIDM00989;BFTC-905 |
| 522 | 191026_b36-t3-8_00di6_00kt6_m04_s_1 | P04 | 2019-10-26 | M04 | BFTC-905 | SIDM00989 | Bladder | Bladder Carcinoma | Urothelial carcinoma | SIDM00989;BFTC-905 |
| 523 | 191026_b61-t2-1_00dsu_00ktw_m06_s_1 | P04 | 2019-10-26 | M06 | BFTC-905 | SIDM00989 | Bladder | Bladder Carcinoma | Urothelial carcinoma | SIDM00989;BFTC-905 |
| 524 | 191125_b32-t4-8_00dge_00n1d_m03_s_1 | P04 | 2019-11-25 | M03 | BFTC-905 | SIDM00989 | Bladder | Bladder Carcinoma | Urothelial carcinoma | SIDM00989;BFTC-905 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 6590 | 200131_b4-7-t3-1_00q3n_00rtc_m05_s_1 | P06 | 2020-01-31 | M05 | COR-L95 | SIDM00521 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00521;COR-L95 |
| 6591 | 200131_b4-9-t3-1_00q3p_00rte_m05_s_1 | P06 | 2020-01-31 | M05 | NCI-H510A | SIDM00927 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00927;NCI-H510A |
| 6592 | 200131_b4-10-t3-1_00q3q_00rtf_m05_s_1 | P06 | 2020-01-31 | M05 | NCI-H2171 | SIDM00733 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00733;NCI-H2171 |
| 6593 | 200131_b4-13-t3-1_00q3t_00rti_m05_s_1 | P06 | 2020-01-31 | M05 | NCI-H1836 | SIDM00770 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00770;NCI-H1836 |
| 6594 | 200201_b3-13-t3-2_00q3d_00rtm_m05_s_1 | P06 | 2020-02-01 | M05 | IST-SL1 | SIDM00223 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00223;IST-SL1 |
1242 rows × 10 columns
Tissue_type¶
[3]:
Tissue_type_counts = metadata['Tissue_type'].value_counts().reset_index()
Tissue_type_counts.columns = ['Tissue_type', 'Count']
Tissue_type_counts
[3]:
| Tissue_type | Count | |
|---|---|---|
| 0 | Lung | 273 |
| 1 | Large Intestine | 271 |
| 2 | Kidney | 181 |
| 3 | Stomach | 153 |
| 4 | Pancreas | 115 |
| 5 | Bladder | 96 |
| 6 | Central Nervous System | 78 |
| 7 | Liver | 75 |
[4]:
fig = px.pie(Tissue_type_counts, values='Count', names='Tissue_type', title="Diverse Tissue Type")
fig.update_traces(textinfo='label+percent', insidetextorientation='radial')
# Save figure to PDF
pio.write_image(fig, 'figs/Tissue_type.pdf')
Cancer_type¶
[5]:
Cancer_type_counts = metadata['Cancer_type'].value_counts().reset_index()
Cancer_type_counts.columns = ['Cancer_type', 'Count']
Cancer_type_counts
[5]:
| Cancer_type | Count | |
|---|---|---|
| 0 | Colorectal Carcinoma | 271 |
| 1 | Small Cell Lung Carcinoma | 187 |
| 2 | Kidney Carcinoma | 181 |
| 3 | Gastric Carcinoma | 153 |
| 4 | Pancreatic Carcinoma | 110 |
| 5 | Bladder Carcinoma | 96 |
| 6 | Non-Small Cell Lung Carcinoma | 86 |
| 7 | Glioma | 78 |
| 8 | Hepatocellular Carcinoma | 75 |
| 9 | Other Solid Carcinomas | 5 |
[6]:
fig = px.pie(Cancer_type_counts, values='Count', names='Cancer_type', title="Diverse Cancer Type")
# Update the labels to show both count and percentage
fig.update_traces(textinfo='label+percent', insidetextorientation='radial')
# Save figure to PDF
pio.write_image(fig, 'figs/Cancer_types.pdf')
Cancer_subtype¶
[7]:
Cancer_subtype = metadata['Cancer_subtype'].value_counts().reset_index()
Cancer_subtype.columns = ['Cancer_subtype', 'Count']
Cancer_subtype["Cancer_subtype_modify"] = [
x if count > 14 else "Others"
for x, count in zip(Cancer_subtype["Cancer_subtype"], Cancer_subtype["Count"])
]
Cancer_subtype
[7]:
| Cancer_subtype | Count | Cancer_subtype_modify | |
|---|---|---|---|
| 0 | Small cell lung carcinoma | 187 | Small cell lung carcinoma |
| 1 | Colon adenocarcinoma | 147 | Colon adenocarcinoma |
| 2 | Kidney carcinoma | 107 | Kidney carcinoma |
| 3 | Bladder carcinoma | 89 | Bladder carcinoma |
| 4 | Squamous cell lung carcinoma | 86 | Squamous cell lung carcinoma |
| 5 | Gastric adenocarcinoma | 75 | Gastric adenocarcinoma |
| 6 | Hepatocellular carcinoma | 72 | Hepatocellular carcinoma |
| 7 | Low grade glioma | 72 | Low grade glioma |
| 8 | Clear cell renal cell carcinoma | 64 | Clear cell renal cell carcinoma |
| 9 | Pancreatic ductal adenocarcinoma | 52 | Pancreatic ductal adenocarcinoma |
| 10 | Cecum adenocarcinoma | 46 | Cecum adenocarcinoma |
| 11 | Pancreatic adenocarcinoma | 44 | Pancreatic adenocarcinoma |
| 12 | Colorectal carcinoma | 43 | Colorectal carcinoma |
| 13 | Rectal adenocarcinoma | 35 | Rectal adenocarcinoma |
| 14 | Gastric signet ring cell adenocarcinoma | 29 | Gastric signet ring cell adenocarcinoma |
| 15 | Gastric tubular adenocarcinoma | 15 | Gastric tubular adenocarcinoma |
| 16 | Pancreatic carcinoma | 13 | Others |
| 17 | Gastric carcinoma | 12 | Others |
| 18 | Urothelial carcinoma | 7 | Others |
| 19 | Gastric fundus carcinoma | 7 | Others |
| 20 | Papillary renal cell carcinoma | 6 | Others |
| 21 | Oligodendroglioma | 6 | Others |
| 22 | Gastic small cell neuroendocrine carcinoma | 6 | Others |
| 23 | Gastric small cell carcinoma | 6 | Others |
| 24 | Pancreatic somatostatinoma | 5 | Others |
| 25 | Renal pelvis and ureter urothelial carcinoma | 4 | Others |
| 26 | Hepatoblastoma | 3 | Others |
| 27 | Gastric choriocarcinoma | 3 | Others |
| 28 | Pancreatic adenosquamous carcinoma | 1 | Others |
[8]:
fig = px.pie(Cancer_subtype, values='Count', names='Cancer_subtype_modify', title="Diverse Cancer subtype")
# Update the labels to show both count and percentage
fig.update_traces(textinfo='label+percent', insidetextorientation='radial')
# Save figure to PDF
pio.write_image(fig, 'figs/Diverse Cancer subtype modify.pdf')
The Sankey diagram delineates the relationships¶
[9]:
def aggregate_by_sum(df, column_name, threshold=14):
# Group by the specified column and calculate the sum for each group
group_counts = df.groupby(column_name).size()
# Select groups with a sum greater than the threshold
processed_data = group_counts[group_counts > threshold].index.tolist()
# Filter the original DataFrame for rows belonging to the filtered groups
processed_data = df[df[column_name].isin(processed_data)]
return processed_data
# 使用函数
result_df = aggregate_by_sum(metadata, 'Cancer_subtype', 10)
result_df
[9]:
| Automatic_MS_filename | Batch | Date | Instrument | Cell_line | SIDM | Tissue_type | Cancer_type | Cancer_subtype | Project_Identifier | |
|---|---|---|---|---|---|---|---|---|---|---|
| 527 | 181010_e0022_p02_2178_1_s_m04_1 | P02 | 2018-10-10 | M04 | SW1710 | SIDM00420 | Bladder | Bladder Carcinoma | Bladder carcinoma | SIDM00420;SW1710 |
| 528 | 181010_e0022_p02_2178_3_s_m04_1 | P02 | 2018-10-10 | M04 | SW1710 | SIDM00420 | Bladder | Bladder Carcinoma | Bladder carcinoma | SIDM00420;SW1710 |
| 529 | 181010_e0022_p02_2178_2_s_m04_1 | P02 | 2018-10-10 | M04 | SW1710 | SIDM00420 | Bladder | Bladder Carcinoma | Bladder carcinoma | SIDM00420;SW1710 |
| 530 | 181127_e0022_p02_2051_3_s_m04_1 | P02 | 2018-11-27 | M04 | RT-112 | SIDM00402 | Bladder | Bladder Carcinoma | Bladder carcinoma | SIDM00402;RT-112 |
| 531 | 181127_e0022_p02_2051_1_s_m04_1 | P02 | 2018-11-27 | M04 | RT-112 | SIDM00402 | Bladder | Bladder Carcinoma | Bladder carcinoma | SIDM00402;RT-112 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 6590 | 200131_b4-7-t3-1_00q3n_00rtc_m05_s_1 | P06 | 2020-01-31 | M05 | COR-L95 | SIDM00521 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00521;COR-L95 |
| 6591 | 200131_b4-9-t3-1_00q3p_00rte_m05_s_1 | P06 | 2020-01-31 | M05 | NCI-H510A | SIDM00927 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00927;NCI-H510A |
| 6592 | 200131_b4-10-t3-1_00q3q_00rtf_m05_s_1 | P06 | 2020-01-31 | M05 | NCI-H2171 | SIDM00733 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00733;NCI-H2171 |
| 6593 | 200131_b4-13-t3-1_00q3t_00rti_m05_s_1 | P06 | 2020-01-31 | M05 | NCI-H1836 | SIDM00770 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00770;NCI-H1836 |
| 6594 | 200201_b3-13-t3-2_00q3d_00rtm_m05_s_1 | P06 | 2020-02-01 | M05 | IST-SL1 | SIDM00223 | Lung | Small Cell Lung Carcinoma | Small cell lung carcinoma | SIDM00223;IST-SL1 |
1188 rows × 10 columns
[10]:
def generate_color_map(labels, alpha_nodes=1.0, alpha_links=0.55):
hues = [i/len(labels) for i in range(len(labels))]
colors_hsv = [(h, 0.5, 0.8) for h in hues]
colors_rgba_nodes = [colorsys.hsv_to_rgb(*hsv) for hsv in colors_hsv]
colors_rgba_links = [(r, g, b, alpha_links) for r, g, b in colors_rgba_nodes]
colors_rgba_nodes = [(r, g, b, alpha_nodes) for r, g, b in colors_rgba_nodes]
colors_str_nodes = ["rgba({},{},{},{})".format(int(r*255), int(g*255), int(b*255), a) for r, g, b, a in colors_rgba_nodes]
colors_str_links = ["rgba({},{},{},{})".format(int(r*255), int(g*255), int(b*255), a) for r, g, b, a in colors_rgba_links]
return dict(zip(labels, colors_str_nodes)), dict(zip(labels, colors_str_links))
def draw_sankey(data, filename=None):
all_labels = sorted(list(set(data['Tissue_type'].unique().tolist() + data['Cancer_type'].unique().tolist() + data['Cancer_subtype'].unique().tolist())))
node_color_map, link_color_map = generate_color_map(all_labels)
# Aggregate data
links = data.groupby(['Tissue_type', 'Cancer_type', 'Cancer_subtype']).size().reset_index(name='count')
final_source = []
final_target = []
final_value = []
for index, row in links.iterrows():
final_source.append(all_labels.index(row['Tissue_type']))
final_target.append(all_labels.index(row['Cancer_type']))
final_value.append(row['count'])
final_source.append(all_labels.index(row['Cancer_type']))
final_target.append(all_labels.index(row['Cancer_subtype']))
final_value.append(row['count'])
link_colors = [link_color_map[all_labels[s]] for s in final_source]
fig = go.Figure(go.Sankey(
node=dict(
pad=15,
thickness=20,
line=dict(color="black", width=0.5),
label=all_labels,
color=[node_color_map[label] for label in all_labels]
),
link=dict(
source=final_source,
target=final_target,
value=final_value,
color=link_colors
)
))
fig.update_layout(title_text="Sankey Diagram of Cancer Types and Subtypes", font_size=10)
if filename:
fig.write_image(filename)
fig.show()
# 使用示例
# df = pd.DataFrame(data)
draw_sankey(result_df, filename="sankey_diagram-3.pdf")
The reproducibility and reliability of the STAVER-processed data¶
HEK293T Spearman correlation analysis¶
[6]:
# Load HEK293T data
HEK293T_rawdata = pd.read_csv("~/STAVER-revised/HEK_293T/HEK-QCS_matrix_rawdata.csv", index_col=0)
HEK293T_rawdata.dropna(thresh=5, inplace=True)
HEK293T_STAVER = pd.read_csv("~/STAVER-revised/HEK_293T/HEK-QCS_matrix_STAVER.csv", index_col=0)
HEK293T_STAVER.dropna(thresh=5, inplace=True)
[26]:
def correlation_heatmap(data, method = 'pearson', log_transformed = False, outpath = None, filename = None):
""" Correaltion heatmap and correlation matrix
Args:
--------------
data -> Dataframe: dataframe of raw data
method -> str: pearson or spearman
log_transformed -> bool: whether to use log10 transformed data
Return:
-----------
Dataframe: Correaltion of experiments data
"""
if log_transformed:
data = np.log10(data)
corr = data.corr(method = method)
## Platelet selection refernce: https://learnku.com/articles/39890
plt.figure(figsize=(10, 10))
# optional cmap: RdYlGn; RdYlGn_r; YlGn; rocket_r; YlGnBu; YlGnBu_r; YlOrBr; YlOrBr_r; YlOrRd; YlOrRd_r; RdBu_r
sns.clustermap(corr, cmap="Reds", annot= False, robust=False, col_cluster=False, row_cluster=False, linewidths=.005,fmt=".2f")
# sn.heatmap(corr, annot=True, cmap='vlag') ## optional cmap: RdYlGn; RdYlGn_r; YlGn; rocket_r; YlGnBu; YlGnBu_r; YlOrBr; YlOrBr_r; YlOrRd; YlOrRd_r;
if outpath:
plt.savefig(f"{outpath}/{filename}_corr_heatmap.pdf")
corr.to_csv(f"{outpath}/{filename}_corr_matrix.csv")
plt.show()
# return corr
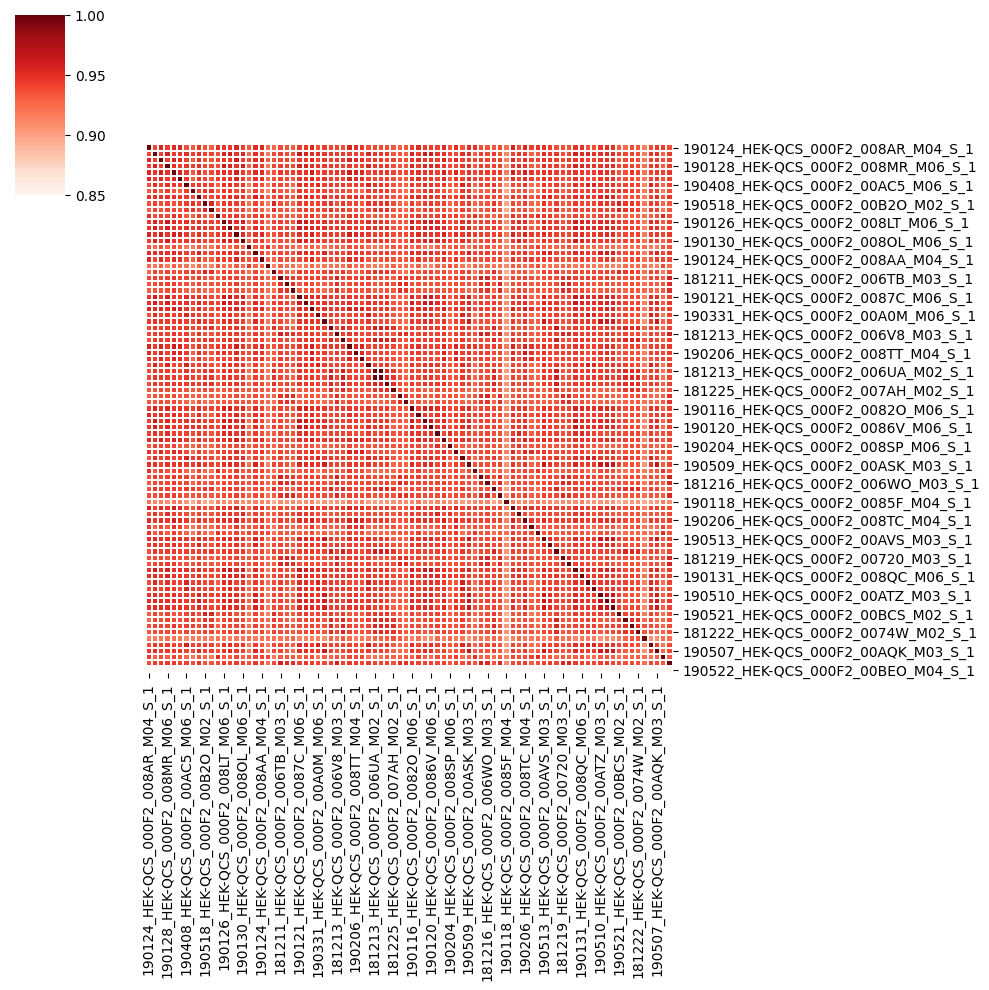
The correlation heatmap of raw data¶
[27]:
outpath = r'~/STAVER-revised/figs/'
correlation_heatmap(HEK293T_rawdata, method = 'spearman', log_transformed=False, outpath = outpath, filename = 'HEK-QCS_matrix_rawdata_corr_heatmap')
<Figure size 1000x1000 with 0 Axes>

The correlation heatmap of STAVER-processed data¶
[28]:
outpath = r'~/STAVER-revised/figs/'
correlation_heatmap(HEK293T_STAVER, method = 'spearman', log_transformed=False, outpath = outpath, filename = 'HEK-QCS_matrix_STAVER_corr_heatmap')
<Figure size 1000x1000 with 0 Axes>
[93]:
def extract_lower_triangular(df_corr):
"""
提取相关性矩阵对角线以下的一半相关性值。
参数:
----
df_corr : pandas.DataFrame
相关性矩阵的 DataFrame。
返回:
-----
pandas.DataFrame
包含对角线以下一半相关性值的 DataFrame。
"""
if not isinstance(df_corr, pd.DataFrame):
raise ValueError("df_corr 必须是 pandas.DataFrame 类型。")
# 获取对角线以下的索引
tril_indices = np.tril_indices(df_corr.shape[0], k=-1)
# 提取对角线以下的一半相关性值
lower_triangular_values = df_corr.to_numpy()[tril_indices]
return pd.DataFrame(
{
'Row': df_corr.index[tril_indices[0]],
'Column': df_corr.columns[tril_indices[1]],
'Correlation': lower_triangular_values,
}
)
def get_IQR(data):
Q1 = data.quantile(0.25)
Q3 = data.quantile(0.75)
IQR = Q3 - Q1
print(f"The IQR1 is: {Q1}")
print(f"The IQR3 is: {Q3}")
mean = data.mean()
print(f"The mean is: {mean}")
median = data.median()
print(f"The median is: {median}")
The IQR of HEK293T spearman correlation matrix in rawdata¶
[105]:
res = extract_lower_triangular(HEK293T_rawdata.corr())
get_IQR(res['Correlation'])
The IQR1 is: 0.8846399725
The IQR3 is: 0.907049235
The mean is: 0.893689345854834
The median is: 0.897238564
The IQR of HEK293T spearman correlation matrix in SATVER-processed data¶
[106]:
res = extract_lower_triangular(HEK293T_STAVER.corr())
get_IQR(res['Correlation'])
The IQR1 is: 0.9321159908757065
The IQR3 is: 0.9821799248475355
The mean is: 0.9523831119684066
The median is: 0.9657469442578246
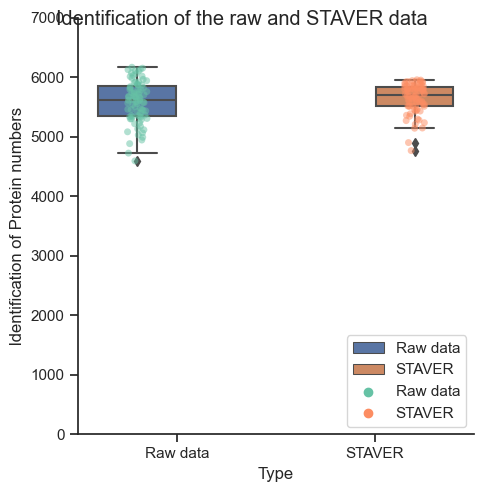
The identification protein numbers of the raw data and STAVER data¶
[2]:
protein_num= pd.read_clipboard()
protein_num
[2]:
| Experiment | ldentification of Protein numbers | Type | |
|---|---|---|---|
| 0 | 190124_HEK-QCS_000F2_008AR_M04_S_1 | 5683 | Raw data |
| 1 | 190124_HEK-QCS_000F2_008IQ_M06_S_1 | 5923 | Raw data |
| 2 | 190125_HEK-QCS_000F2_008JO_M04_S_1 | 5617 | Raw data |
| 3 | 190128_HEK-QCS_000F2_008MR_M06_S_1 | 5873 | Raw data |
| 4 | 190129_HEK-QCS_000F2_008NM_M04_S_1 | 5465 | Raw data |
| ... | ... | ... | ... |
| 162 | 190102_HEK-QCS_000F2_007K6_M02_S_1 | 5150 | STAVER |
| 163 | 190401_HEK-QCS_000F2_00A13_M06_S_1 | 5817 | STAVER |
| 164 | 190507_HEK-QCS_000F2_00AQK_M03_S_1 | 5961 | STAVER |
| 165 | 190522_HEK-QCS_000F2_00BEO_M04_S_1 | 5283 | STAVER |
| 166 | 181213_HEK-QCS_000F2_006UR_M03_S_1 | 5519 | STAVER |
167 rows × 3 columns
[28]:
def plot_protein_numbers(data, x_col, y_col, title, y_lim=None, height=6, aspect=1.3):
"""
Creates a combined box and strip plot for protein number visualization.
Args:
data (pd.DataFrame): DataFrame containing the data to be plotted.
x_col (str): Column name in `data` to be plotted on the x-axis.
y_col (str): Column name in `data` to be plotted on the y-axis.
title (str): Title of the plot.
y_lim (tuple, optional): Tuple specifying the limits for the y-axis (e.g., (0, 7000)).
height (float, optional): Height (in inches) of each facet. Defaults to 6.
aspect (float, optional): Aspect ratio of each facet, so that aspect * height gives the width of each facet in inches. Defaults to 1.3.
Example:
>>> plot_protein_numbers(protein_num, 'Type', 'Identification of Protein numbers',
'Protein numbers of HEK293T QC samples', y_lim=(0, 7000), height=5, aspect=1)
"""
custom_params = {"axes.spines.right": False, "axes.spines.top": False}
sns.set_theme(style="ticks", rc=custom_params)
# Create a box plot
box_plot = sns.catplot(x=x_col, y=y_col, hue=x_col, kind="box", legend=False, height=height, aspect=aspect, data=data)
# Overlay with a strip plot
sns.stripplot(x=x_col, y=y_col, hue=x_col, jitter=True, dodge=True, marker='o', palette="Set2", alpha=0.5, data=data)
# Set additional plot attributes
box_plot.fig.suptitle(title) # Set the title for the figure
plt.legend(loc='lower right')
if y_lim:
plt.ylim(y_lim)
plt.show()
# The protein numbers of the raw data and STAVER data
plot_protein_numbers(protein_num, 'Type', 'ldentification of Protein numbers',
'Identification of the raw and STAVER data', y_lim=(0, 7000), height=5, aspect=1)
The Coefficient of Variation (CVs) of the raw data and STAVER data¶
[130]:
# custom_params = {"axes.spines.right": False, "axes.spines.top": False}
# sns.set_theme(style="ticks", rc=custom_params)
def plot_molecular_variance(df, column_name):
"""
Plots the density curves of the original and STAVER processed data for comparison.
Args:
df: A pandas dataframe containing the data.
column_name: A string representing the column name of the data to be plotted.
Returns:
None
"""
plt.figure(figsize=(5, 4))
# Density plot of the original data
sns.kdeplot(df[df[column_name]=='Raw data']['Coefficient of Variation [%]'], label='Original Density', color='blue', linestyle="--")
# Density plot of the STAVER-processed data
sns.kdeplot(df[df[column_name]=='STAVER']['Coefficient of Variation [%]'], label='STAVER Density', color='red')
plt.legend()
plt.title('Coefficient of Variation density curve')
plt.xlabel("Coefficient of Variation [%]")
plt.ylabel('Density')
plt.show()
[131]:
protein_cv = pd.read_csv("~/STAVER-revised/Pan-cancer-cell-lines/HEK293T_Protein_CV_Compare.csv")
plot_molecular_variance(protein_cv, 'Type')

The advantages of the STAVER algorithm to uncover inherent biological differences¶
Distinct tumors often exhibit diverse molecular characteristics (Cell, 2014, PMID: 25109877; Cell, 2023, PMID: 37582357), with even different histological subtypes of the same tumor demonstrating significant molecular heterogeneity (Science, 2014, PMID: 25301631; Cancer Cell, 2023, PMID: 36563681), contributing to challenges in cancer treatment. Thus, accurately deciphering the inherent molecular heterogeneity of various cancer types, especially in high-dimensional data such as proteomics, is vital for precision treatments and ultimately improved patient outcomes.
To demonstrate the superior advantages of the STAVER algorithm to uncover inherent biological differences, we comprehensively compared the original data and STAVER-processed data of the 1,242 cancer cell line samples from diverse cancer types.
The UMAP analysis of diverse 1242 cancer cell line samples¶
[98]:
def visualize_cancer_subtypes(proteomics_data_path,
subtypes_file_path,
color_params=['Batch', 'Tissue_type', 'Cancer_type'],
figsize_per_plot=(4.5, 4),
legend_loc="on data",
outpath='./',
filename=None,
add_outline=False,
show_plot=False):
"""
Visualize the given proteomics data and cancer subtypes using t-SNE and UMAP.
Args:
proteomics_data_path (str): Path to the proteomics data (samples as rows, proteins as columns).
subtypes_file_path (str): Path to the file with samples and their corresponding cancer subtypes.
color_params (list): List of parameters to color by in the plots.
figsize_per_plot (tuple): Size of each individual plot.
outpath (str): Directory to save the plots.
filename (str): Prefix of the saved files.
Returns:
None. Generates visualization images for t-SNE and UMAP.
"""
# Load data
data = pd.read_csv(proteomics_data_path, index_col=0).T.replace(np.nan, 0)
# Load subtype information
subtypes = pd.read_csv(subtypes_file_path, index_col=0)
# Check for missing subtype information
if not all(sample in subtypes.index for sample in data.index):
raise ValueError("Some samples lack subtype information!")
# Convert data to AnnData format
adata = anndata.AnnData(X=data)
# Add subtype information to AnnData
adata.obs = subtypes.loc[adata.obs_names]
# Data normalization (if required)
sc.pp.scale(adata)
# Compute t-SNE and UMAP
sc.tl.tsne(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
# Set filenames if not provided
if not filename:
filename = os.path.basename(proteomics_data_path).split('.')[0]
# # Visualize t-SNE
# fig, axes = plt.subplots(figsize=(len(color_params)*figsize_per_plot[0], figsize_per_plot[1]), nrows=1, ncols=len(color_params))
# for i, param in enumerate(color_params):
# sc.pl.tsne(adata, color=param, legend_loc=legend_loc, title=f"t-SNE colored by {param}", ax=axes[i], show=show_plot)
# fig.tight_layout()
# fig.savefig(os.path.join(outpath, f"{filename}_tsne.pdf"))
# plt.close(fig)
# # Visualize UMAP
# fig, axes = plt.subplots(figsize=(len(color_params)*figsize_per_plot[0], figsize_per_plot[1]), nrows=1, ncols=len(color_params))
# for i, param in enumerate(color_params):
# sc.pl.umap(adata, color=param, legend_loc=legend_loc, title=f"UMAP colored by {param}", ax=axes[i], show=show_plot)
# fig.tight_layout()
# fig.savefig(os.path.join(outpath, f"{filename}_umap.pdf"))
# plt.close(fig)
# Determine the number of plots (number of color_params x 2 for both t-SNE and UMAP)
nrows = 2
ncols = len(color_params)
# Create a figure with the necessary number of subplots
fig, axes = plt.subplots(nrows=nrows, ncols=ncols, figsize=(ncols * figsize_per_plot[0], nrows * figsize_per_plot[1]))
# Visualize t-SNE on the first row
for i, param in enumerate(color_params):
sc.pl.tsne(adata, color=param, legend_loc=legend_loc, title=f"t-SNE colored by {param}", add_outline=add_outline, ax=axes[0, i], show=show_plot)
# Visualize UMAP on the second row
for i, param in enumerate(color_params):
sc.pl.umap(adata, color=param, legend_loc=legend_loc, title=f"UMAP colored by {param}", add_outline=add_outline, ax=axes[1, i], show=show_plot)
# Save the combined visualization
fig.tight_layout()
fig.savefig(os.path.join(outpath, f"{filename}_combined.pdf"))
plt.show()
plt.close(fig)
return adata
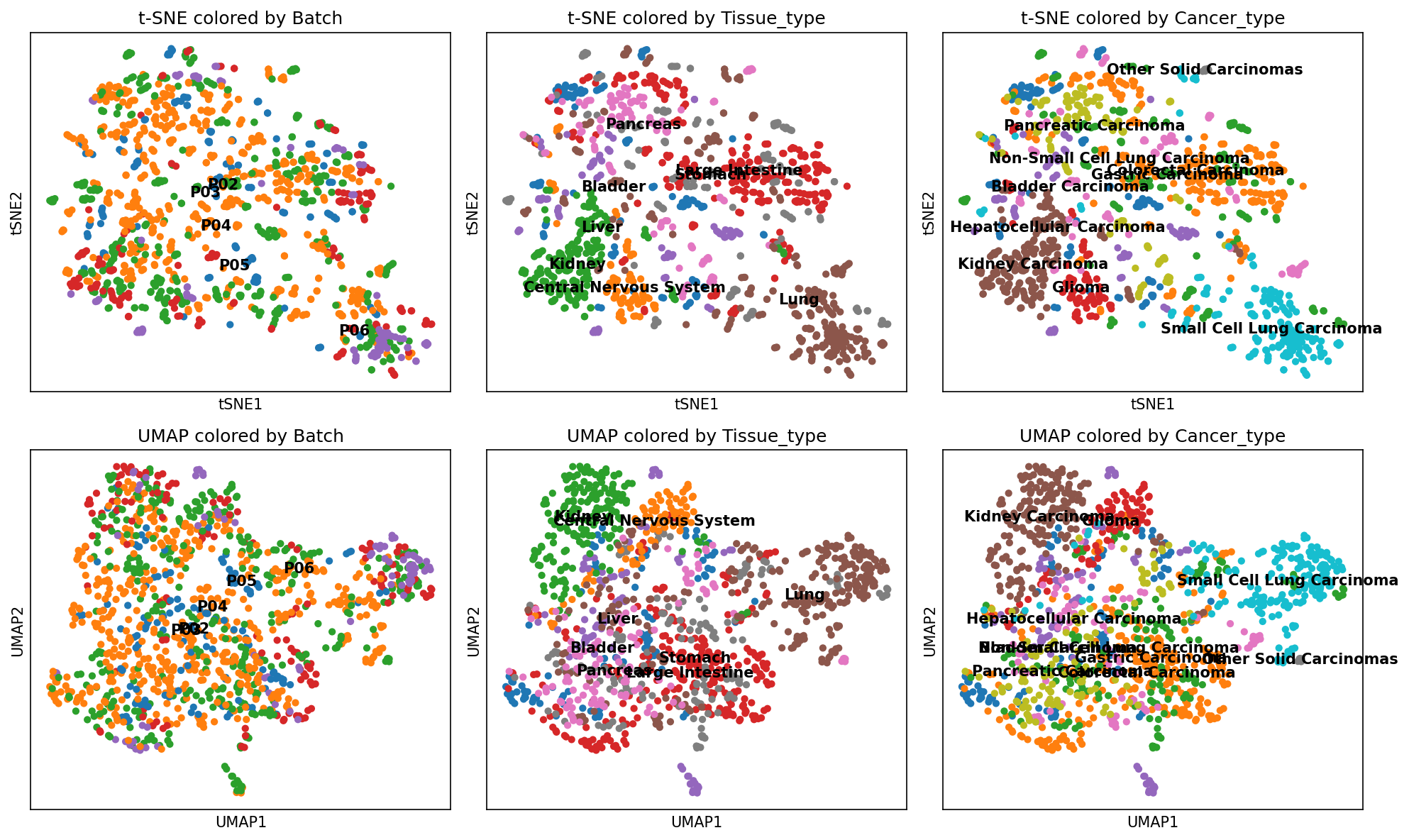
The UMAP analysis of the Rawdata¶
By employing UMAP analysis, the results indicated that the STAVER algorithm did not introduce batch effects, corroborating the original data findings (Figure RL2H-2I and Figure RL5H-5I). In the original data, the UMAP analysis showed that the 1,242 cancer cell line samples were diffusely distributed across various tissue sources and cancer types and did not show a clear separation. This lack of distinction was particularly evidenced among bladder, colorectal, pancreatic, gastric, and hepatocellular cancer cell lines, which intermixed with each other (Figure RL2H and Figure RL5H).
[100]:
outpath = '~/DIA-STAVER/STAVER-nc-revised/revised-sript/figs-1/'
if not os.path.exists(outpath):
os.makedirs(outpath)
adata_rawdata = visualize_cancer_subtypes("~/STAVER-revised/PCA/rawdata_data.csv", "~/STAVER-revised/PCA/metadata_anatation_scanpy.csv", outpath=outpath, filename = "Raw_data_2")
WARNING: You’re trying to run this on 11880 dimensions of `.X`, if you really want this, set `use_rep='X'`.
Falling back to preprocessing with `sc.pp.pca` and default params.
OMP: Info #271: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
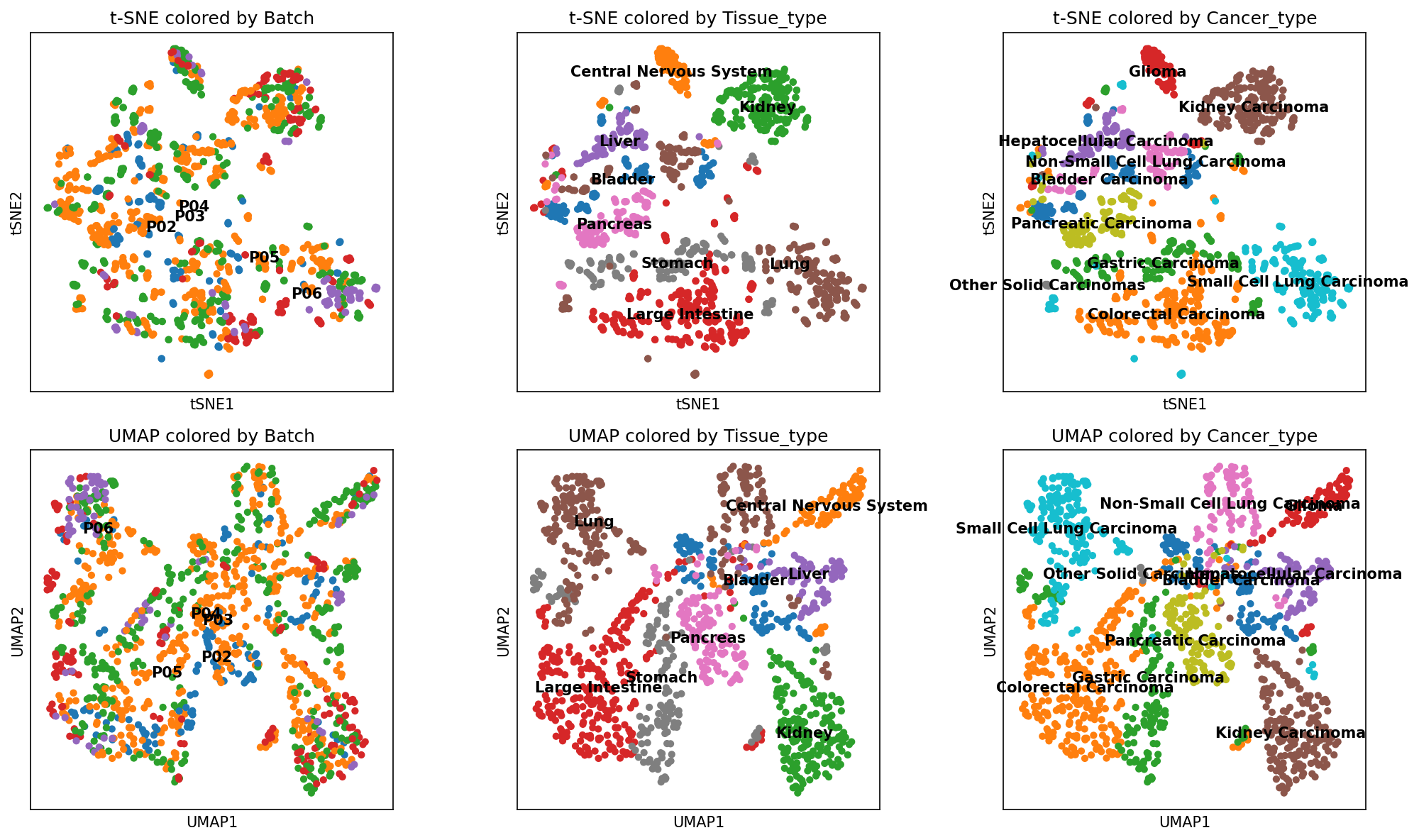
The UMAP analysis of STAVER-processed data¶
When analyzing STAVER-processed data, UMAP visualization clearly separated the 1,242 cancer cell line samples by tissue sources and cancer types (Figure RL2I and Figure RL5I). Notably, the STAVER-processed data revealed that cancer cell lines from the digestive tract, such as intestinal, gastric, and pancreatic, exhibited more molecular similarities than their nondigestive tract counterparts (lung, glioma, and kidney cancer cell lines). These findings highlighted the potential superiority of the STAVER algorithm in elucidating the intrinsic biological differences among diverse tumor cell lines.
[102]:
outpath = '~/DIA-STAVER/STAVER-nc-revised/revised-sript/figs-1/'
if not os.path.exists(outpath):
os.makedirs(outpath)
adata_STAVER = visualize_cancer_subtypes("~/STAVER-revised/PCA/STAVER_data.csv", "~/STAVER-revised/PCA/metadata_anatation_scanpy.csv", outpath=outpath, filename = "STAVER_data_2")
WARNING: You’re trying to run this on 8202 dimensions of `.X`, if you really want this, set `use_rep='X'`.
Falling back to preprocessing with `sc.pp.pca` and default params.
Cancer specific proteins¶
To rigorously establish the advantages of the STAVER algorithm in identifying tumor cell-specific proteins, we compared the distribution patterns of cell-specific protein expression profiles between the original and STAVER-processed data. We incorporated the Human Protein Atlas (HPA) molecular annotations of tumor cell specificity and their expression profiles across various cancer cell lines for integrated analysis.
The results demonstrated that the STAVER-processed data more accurately reflected cancer type-specific expression patterns than the original proteomic data.
[4]:
HPA_dataset = pd.read_csv("~/STAVER-nc-revised/resources/proteinatlas_56a92855.tsv", sep="\t")
HPA_dataset
[4]:
| Gene | Gene synonym | Ensembl | Gene description | Uniprot | Chromosome | Position | Protein class | Biological process | Molecular function | ... | Pathology prognostics - Lung cancer | Pathology prognostics - Melanoma | Pathology prognostics - Ovarian cancer | Pathology prognostics - Pancreatic cancer | Pathology prognostics - Prostate cancer | Pathology prognostics - Renal cancer | Pathology prognostics - Stomach cancer | Pathology prognostics - Testis cancer | Pathology prognostics - Thyroid cancer | Pathology prognostics - Urothelial cancer | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | A1BG | NaN | ENSG00000121410 | Alpha-1-B glycoprotein | P04217 | 19 | 58345178-58353492 | Plasma proteins, Predicted intracellular prote... | NaN | NaN | ... | unprognostic (1.09e-1) | unprognostic (2.59e-1) | unprognostic (2.10e-1) | unprognostic (1.47e-2) | unprognostic (1.37e-2) | unprognostic (4.19e-5) | unprognostic (2.37e-2) | unprognostic (1.94e-1) | unprognostic (1.72e-1) | unprognostic (6.72e-2) |
| 1 | A1CF | ACF, ACF64, ACF65, APOBEC1CF, ASP | ENSG00000148584 | APOBEC1 complementation factor | Q9NQ94 | 10 | 50799409-50885675 | Predicted intracellular proteins | mRNA processing | RNA-binding | ... | unprognostic (7.38e-3) | NaN | unprognostic (1.30e-2) | unprognostic (2.46e-2) | unprognostic (1.20e-1) | unprognostic (1.90e-3) | unprognostic (1.97e-2) | unprognostic (2.77e-1) | unprognostic (2.19e-2) | unprognostic (8.50e-4) |
| 2 | A2M | CPAMD5, FWP007, S863-7 | ENSG00000175899 | Alpha-2-macroglobulin | P01023 | 12 | 9067664-9116229 | Cancer-related genes, Candidate cardiovascular... | NaN | Protease inhibitor, Serine protease inhibitor | ... | unprognostic (3.65e-2) | unprognostic (2.38e-1) | unprognostic (7.19e-2) | unprognostic (4.71e-2) | unprognostic (2.06e-2) | unprognostic (1.28e-2) | unprognostic (8.04e-3) | unprognostic (2.32e-2) | unprognostic (8.58e-2) | unprognostic (9.03e-3) |
| 3 | A2ML1 | CPAMD9, FLJ25179, p170 | ENSG00000166535 | Alpha-2-macroglobulin like 1 | A8K2U0 | 12 | 8822621-8887001 | Disease related genes, Predicted intracellular... | NaN | Protease inhibitor, Serine protease inhibitor | ... | unprognostic (7.58e-3) | unprognostic (2.63e-1) | unprognostic (1.57e-1) | unprognostic (1.15e-3) | unprognostic (2.03e-1) | unprognostic (1.06e-9) | unprognostic (2.28e-1) | unprognostic (3.07e-1) | unprognostic (5.88e-2) | unprognostic (2.42e-2) |
| 4 | A3GALT2 | A3GALT2P, IGB3S, IGBS3S | ENSG00000184389 | Alpha 1,3-galactosyltransferase 2 | U3KPV4 | 1 | 33306766-33321098 | Enzymes, Predicted membrane proteins | Lipid metabolism | Glycosyltransferase, Transferase | ... | unprognostic (4.96e-2) | unprognostic (6.83e-2) | unprognostic (5.81e-2) | unprognostic (1.23e-1) | unprognostic (1.89e-1) | unprognostic (4.90e-8) | unprognostic (1.17e-1) | NaN | unprognostic (1.12e-2) | unprognostic (7.87e-2) |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 20157 | ZYG11A | ZYG11 | ENSG00000203995 | Zyg-11 family member A, cell cycle regulator | Q6WRX3 | 1 | 52842511-52894998 | Predicted intracellular proteins | Ubl conjugation pathway | NaN | ... | unprognostic (2.34e-1) | unprognostic (4.56e-2) | unprognostic (2.06e-2) | unprognostic (4.01e-2) | unprognostic (1.01e-1) | unprognostic (6.15e-3) | unprognostic (2.95e-1) | unprognostic (1.21e-1) | unprognostic (3.07e-1) | unprognostic (1.02e-1) |
| 20158 | ZYG11B | FLJ13456, ZYG11 | ENSG00000162378 | Zyg-11 family member B, cell cycle regulator | Q9C0D3 | 1 | 52726453-52827336 | Predicted intracellular proteins | Ubl conjugation pathway | NaN | ... | unprognostic (1.85e-1) | unprognostic (4.84e-3) | unprognostic (5.06e-2) | unprognostic (2.76e-1) | unprognostic (6.08e-2) | prognostic favorable (9.80e-7) | unprognostic (2.22e-1) | unprognostic (3.37e-1) | unprognostic (1.13e-1) | unprognostic (9.57e-2) |
| 20159 | ZYX | NaN | ENSG00000159840 | Zyxin | Q15942 | 7 | 143381295-143391111 | Plasma proteins, Predicted intracellular proteins | Cell adhesion, Host-virus interaction | NaN | ... | unprognostic (1.66e-3) | unprognostic (2.60e-1) | unprognostic (4.22e-1) | unprognostic (1.98e-1) | unprognostic (2.43e-1) | prognostic unfavorable (7.92e-5) | unprognostic (1.39e-1) | unprognostic (8.12e-2) | unprognostic (1.95e-1) | unprognostic (6.72e-2) |
| 20160 | ZZEF1 | FLJ10821, KIAA0399, ZZZ4 | ENSG00000074755 | Zinc finger ZZ-type and EF-hand domain contain... | O43149 | 17 | 4004445-4143030 | Predicted membrane proteins | Transcription, Transcription regulation | Activator | ... | unprognostic (1.44e-2) | unprognostic (1.23e-1) | unprognostic (2.21e-2) | prognostic favorable (6.08e-4) | unprognostic (1.54e-1) | unprognostic (1.38e-3) | unprognostic (6.09e-3) | unprognostic (1.80e-1) | unprognostic (9.43e-2) | unprognostic (6.46e-2) |
| 20161 | ZZZ3 | ATAC1, DKFZP564I052 | ENSG00000036549 | Zinc finger ZZ-type containing 3 | Q8IYH5 | 1 | 77562416-77683419 | Predicted intracellular proteins | Transcription, Transcription regulation | DNA-binding | ... | unprognostic (2.91e-1) | unprognostic (9.67e-2) | unprognostic (1.35e-1) | unprognostic (2.83e-1) | unprognostic (1.91e-1) | unprognostic (1.71e-1) | unprognostic (3.96e-1) | unprognostic (1.95e-1) | unprognostic (3.05e-2) | unprognostic (1.43e-1) |
20162 rows × 89 columns
[6]:
def get_hpa_expression_profile(protein):
HPA = HPA_dataset[['Gene', 'RNA cell line specific nTPM']]
HPA.set_index('Gene', inplace=True)
return HPA.loc[protein, "RNA cell line specific nTPM"]
CTSE overexpression in Gastric, pancreatic, and colorectal cancer of HPA dataset¶
[9]:
get_hpa_expression_profile("CTSE")
[9]:
'colorectal cancer: 28.4;Esophageal cancer: 48.8;Gastric cancer: 110.7;pancreatic cancer: 35.3'
[10]:
HPA_protein_CTSE = {
'Bladder Carcinoma': 0,
'Colorectal Carcinoma': 28.4,
'Gastric Carcinoma': 110.7,
'Glioma': 0,
'Hepatocellular Carcinoma': 0,
'Kidney Carcinoma': 0,
'NSCLC': 0,
'Pancreatic Carcinoma': 35.3,
'SCLC': 0
}
HPA_protein_CTSE = pd.DataFrame.from_dict(HPA_protein_CTSE, orient='index', columns=['CTSE'])
HPA_protein_CTSE = HPA_protein_CTSE.sort_values(by=['CTSE'], ascending=False)
HPA_protein_CTSE
[10]:
| CTSE | |
|---|---|
| Gastric Carcinoma | 110.7 |
| Pancreatic Carcinoma | 35.3 |
| Colorectal Carcinoma | 28.4 |
| Bladder Carcinoma | 0.0 |
| Glioma | 0.0 |
| Hepatocellular Carcinoma | 0.0 |
| Kidney Carcinoma | 0.0 |
| NSCLC | 0.0 |
| SCLC | 0.0 |
[11]:
STAVER = pd.read_csv("~/STAVER-revised/model-evlauate/STAVER_data.csv", index_col=0)
raw_data = pd.read_csv("~/STAVER-revised/model-evlauate/raw_data.csv", index_col=0)
[12]:
def process_data(data, target_gene):
df = data[[target_gene, 'Cancer_type']]
df.replace(0, np.nan, inplace=True)
df.drop(df[df['Cancer_type'] == 'Other Solid Carcinomas'].index, inplace=True)
df = df.groupby('Cancer_type').median()
df = df.sort_values(by=[target_gene], ascending=False)
return df
[13]:
rawdata_CTSE = process_data(raw_data, "CTSE")
rawdata_CTSE.sort_values(by=['CTSE'], ascending=False, inplace=True)
rawdata_CTSE
[13]:
| CTSE | |
|---|---|
| Cancer_type | |
| Gastric Carcinoma | 13.064867 |
| Pancreatic Carcinoma | 13.046290 |
| Hepatocellular Carcinoma | 12.683957 |
| Colorectal Carcinoma | 11.875120 |
| Non-Small Cell Lung Carcinoma | 11.738225 |
| Bladder Carcinoma | 11.182161 |
| Small Cell Lung Carcinoma | 10.270660 |
| Kidney Carcinoma | 10.095740 |
| Glioma | NaN |
[17]:
STAVER_CTSE = process_data(STAVER, "CTSE")
STAVER_CTSE.sort_values(by=['CTSE'], ascending=False, inplace=True)
STAVER_CTSE
[17]:
| CTSE | |
|---|---|
| Cancer_type | |
| Gastric Carcinoma | 9.501038 |
| Pancreatic Carcinoma | 8.500000 |
| Colorectal Carcinoma | 8.050000 |
| Bladder Carcinoma | NaN |
| Glioma | NaN |
| Hepatocellular Carcinoma | NaN |
| Kidney Carcinoma | NaN |
| Non-Small Cell Lung Carcinoma | NaN |
| Small Cell Lung Carcinoma | NaN |
[ ]:
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams['figure.dpi']= 150 # 一般将dpi设置在150到300之间
custom_params = {"axes.spines.right": False, "axes.spines.top": False}
sns.set_theme(style="ticks", rc=custom_params)
def plot_barplot(df, target_col, log_transform=False, x_label=None, y_label=None, title=None, save_path=None):
"""
Plots a barplot.
Args:
df (pd.DataFrame): The dataframe.
target_col (str): The column name to be plotted as the target column.
x_label (str): The x-axis label. Default is None.
y_label (str): The y-axis label. Default is None.
title (str): The title of the plot. Default is None.
save_path (str): The path to save the image. Default is None, indicating no saving.
Returns:
None
"""
plt.figure(figsize=(4, 2.7))
if log_transform:
sns.barplot(x=df.index, y=np.log2(df[target_col]+1))
# Use seaborn's barplot function to plot the graph
else:
sns.barplot(x=df.index, y=df[target_col])
# Set the title and axis labels
if title:
plt.title(title)
if x_label:
plt.xlabel(x_label)
else:
plt.xlabel("Index")
if y_label:
plt.ylabel(y_label)
else:
plt.ylabel(target_col)
plt.xticks(rotation=30, ha='right') # Rotate the x-axis labels for better display
# Save the image (if save_path is specified)
if save_path:
plt.savefig(save_path, bbox_inches='tight')
# Show the image
plt.show()
[33]:
import matplotlib.pyplot as plt
import seaborn as sns
import numpy as np
# 全局的绘图参数设置
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams['figure.dpi'] = 150
custom_params = {"axes.spines.right": False, "axes.spines.top": False}
sns.set_theme(style="ticks", rc=custom_params)
def plot_combined_barplots(datasets, target_col, titles, log_transforms, x_labels, y_labels, save_path=None):
"""
Plots multiple barplots on a single figure, one for each dataset provided.
"""
if not all(len(lst) == len(datasets) for lst in [titles, log_transforms, x_labels, y_labels]):
raise ValueError("All list arguments must be of the same length as the 'datasets' list.")
n = len(datasets)
fig, axes = plt.subplots(1, n, figsize=(n * 4, 4.2))
if n == 1: # If there is only one dataset, axes will not be an array
axes = [axes]
for i, (df, title, log_transform, x_label, y_label) in enumerate(zip(datasets, titles, log_transforms, x_labels, y_labels)):
ax = axes[i]
plot_data = np.log2(df[target_col]+1) if log_transform else df[target_col]
sns.barplot(ax=ax, x=df.index, y=plot_data)
ax.set_title(title)
ax.set_xlabel(x_label if x_label else "Index")
ax.set_ylabel(y_label if y_label else target_col)
# Set the tick parameters and label rotation
ax.set_xticklabels(ax.get_xticklabels(), rotation=30, ha='right')
# Adjust the left margin if needed
plt.subplots_adjust(left=0.1) # You might need to tweak this value
if save_path:
plt.savefig(save_path, bbox_inches='tight')
plt.tight_layout()
plt.show()
# The barplot of CTSE expression profiles
plot_combined_barplots(
datasets=[HPA_protein_CTSE, rawdata_CTSE, STAVER_CTSE],
target_col="CTSE",
titles=["Expression profiles from HPA dataset (CTSE)",
"Expression profiles from original data (CTSE)",
"Expression profiles from STAVER data (CTSE)"],
log_transforms=[True, False, False],
x_labels=[None, None, None],
y_labels=["Log2 (Relative expression)",
"Log2 (Relative expression)",
"Log2 (Relative expression)"],
save_path="combined_barplots_CTSE.pdf"
)
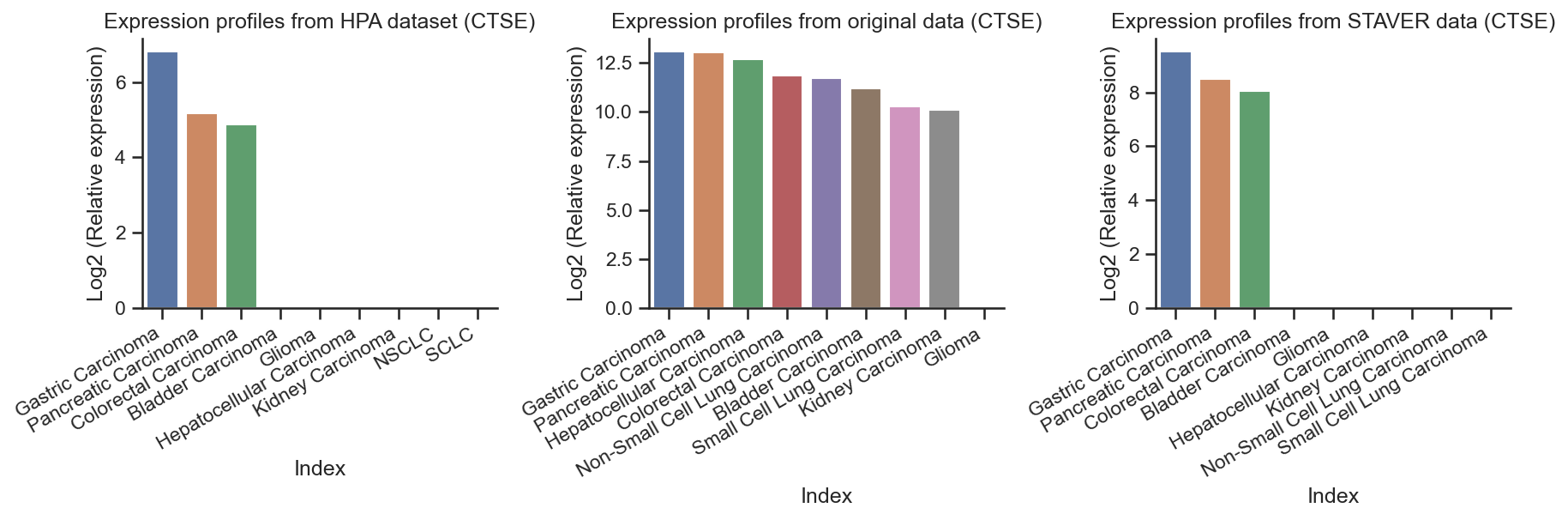
[ ]:
# The barplot of CTSE expression profiles
plot_combined_barplots(
datasets=[HPA_protein_CTSE, rawdata_CTSE, STAVER_CTSE],
target_col="CTSE",
titles=["Expression profiles from HPA dataset (CTSE)",
"Expression profiles from original data (CTSE)",
"Expression profiles from STAVER data (CTSE)"],
log_transforms=[True, False, False],
x_labels=[None, None, None],
y_labels=["Log2 (Relative expression)",
"Log2 (Relative expression)",
"Log2 (Relative expression)"],
save_path="combined_barplots_CTSE.pdf"
)
GPA33 overexpression in colorectal cancer and Gastric cancer of HPA dataset¶
[34]:
get_hpa_expression_profile("GPA33")
[34]:
'Bile duct cancer: 34.1;colorectal cancer: 100.5;Gastric cancer: 19.1'
[35]:
HPA_protein_CTSE = {
'Bladder Carcinoma': 0,
'Colorectal Carcinoma': 100.5,
'Gastric Carcinoma': 19.1,
'Glioma': 0,
'Hepatocellular Carcinoma': 0,
'Kidney Carcinoma': 0,
'NSCLC': 0,
'Pancreatic Carcinoma': 0,
'SCLC': 0
}
HPA_protein_GPA33 = pd.DataFrame.from_dict(HPA_protein_CTSE, orient='index', columns=['GPA33'])
HPA_protein_GPA33 = HPA_protein_GPA33.sort_values(by=['GPA33'], ascending=False)
HPA_protein_GPA33
[35]:
| GPA33 | |
|---|---|
| Colorectal Carcinoma | 100.5 |
| Gastric Carcinoma | 19.1 |
| Bladder Carcinoma | 0.0 |
| Glioma | 0.0 |
| Hepatocellular Carcinoma | 0.0 |
| Kidney Carcinoma | 0.0 |
| NSCLC | 0.0 |
| Pancreatic Carcinoma | 0.0 |
| SCLC | 0.0 |
[39]:
rawdata_GPA33 = process_data(raw_data, "GPA33")
rawdata_GPA33.sort_values(by=['GPA33'], ascending=False, inplace=True)
rawdata_GPA33
[39]:
| GPA33 | |
|---|---|
| Cancer_type | |
| Colorectal Carcinoma | 16.132530 |
| Gastric Carcinoma | 14.659133 |
| Non-Small Cell Lung Carcinoma | 13.211101 |
| Pancreatic Carcinoma | 10.548243 |
| Hepatocellular Carcinoma | 9.851693 |
| Kidney Carcinoma | 9.565263 |
| Bladder Carcinoma | 9.219547 |
| Small Cell Lung Carcinoma | 9.125549 |
| Glioma | 9.036120 |
[41]:
STAVER_GPA33 = process_data(STAVER, "GPA33")
STAVER_GPA33.sort_values(by=['GPA33'], ascending=False, inplace=True)
STAVER_GPA33
[41]:
| GPA33 | |
|---|---|
| Cancer_type | |
| Colorectal Carcinoma | 9.632096 |
| Gastric Carcinoma | 7.820000 |
| Bladder Carcinoma | NaN |
| Glioma | NaN |
| Hepatocellular Carcinoma | NaN |
| Kidney Carcinoma | NaN |
| Non-Small Cell Lung Carcinoma | NaN |
| Pancreatic Carcinoma | NaN |
| Small Cell Lung Carcinoma | NaN |
[42]:
# The barplot of GPA33 expression profiles
plot_combined_barplots(
datasets=[HPA_protein_GPA33, rawdata_GPA33, STAVER_GPA33],
target_col="GPA33",
titles=["Expression profiles from HPA dataset (GPA33)",
"Expression profiles from original data (GPA33)",
"Expression profiles from STAVER data (GPA33)"],
log_transforms=[True, False, False],
x_labels=[None, None, None],
y_labels=["Log2 (Relative expression)",
"Log2 (Relative expression)",
"Log2 (Relative expression)"],
save_path="combined_barplots_GPA33.pdf"
)
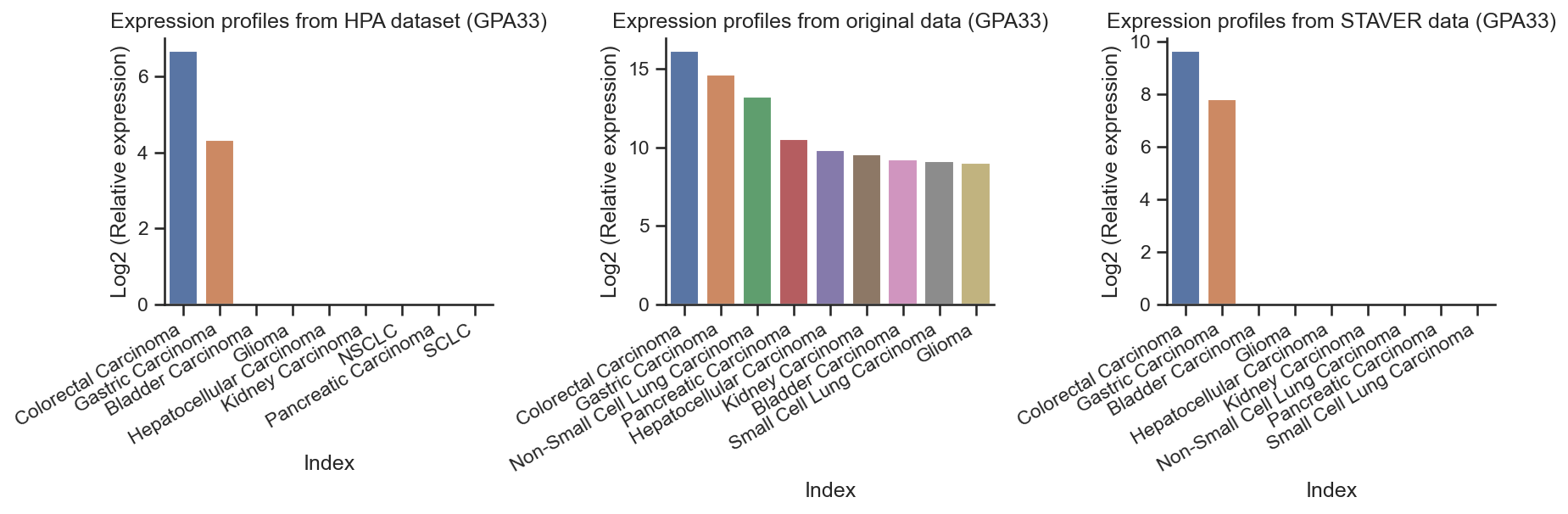
ADGRF1 overexpression in pancreatic cancer of HPA dataset¶
[43]:
get_hpa_expression_profile("ADGRF1")
[43]:
'pancreatic cancer: 62.2'
[44]:
HPA_protein_CTSE = {
'Bladder Carcinoma': 0,
'Colorectal Carcinoma': 0,
'Gastric Carcinoma': 0,
'Glioma': 0,
'Hepatocellular Carcinoma': 0,
'Kidney Carcinoma': 0,
'NSCLC': 0,
'Pancreatic Carcinoma': 62.2,
'SCLC': 0
}
HPA_protein_ADGRF1 = pd.DataFrame.from_dict(HPA_protein_CTSE, orient='index', columns=['ADGRF1'])
HPA_protein_ADGRF1 = HPA_protein_ADGRF1.sort_values(by=['ADGRF1'], ascending=False)
HPA_protein_ADGRF1
[44]:
| ADGRF1 | |
|---|---|
| Pancreatic Carcinoma | 62.2 |
| Bladder Carcinoma | 0.0 |
| Colorectal Carcinoma | 0.0 |
| Gastric Carcinoma | 0.0 |
| Glioma | 0.0 |
| Hepatocellular Carcinoma | 0.0 |
| Kidney Carcinoma | 0.0 |
| NSCLC | 0.0 |
| SCLC | 0.0 |
[45]:
rawdata_ADGRF1 = process_data(raw_data, "ADGRF1")
rawdata_ADGRF1.sort_values(by=['ADGRF1'], ascending=False, inplace=True)
rawdata_ADGRF1
[45]:
| ADGRF1 | |
|---|---|
| Cancer_type | |
| Pancreatic Carcinoma | 10.272240 |
| Gastric Carcinoma | 10.256628 |
| Colorectal Carcinoma | 10.093484 |
| Bladder Carcinoma | 9.910033 |
| Glioma | NaN |
| Hepatocellular Carcinoma | NaN |
| Kidney Carcinoma | NaN |
| Non-Small Cell Lung Carcinoma | NaN |
| Small Cell Lung Carcinoma | NaN |
[46]:
STAVER_ADGRF1 = process_data(STAVER, "ADGRF1")
STAVER_ADGRF1.sort_values(by=['ADGRF1'], ascending=False, inplace=True)
STAVER_ADGRF1
[46]:
| ADGRF1 | |
|---|---|
| Cancer_type | |
| Pancreatic Carcinoma | 7.594326 |
| Bladder Carcinoma | NaN |
| Colorectal Carcinoma | NaN |
| Gastric Carcinoma | NaN |
| Glioma | NaN |
| Hepatocellular Carcinoma | NaN |
| Kidney Carcinoma | NaN |
| Non-Small Cell Lung Carcinoma | NaN |
| Small Cell Lung Carcinoma | NaN |
[47]:
# The barplot of ADGRF1 expression profiles
plot_combined_barplots(
datasets=[HPA_protein_ADGRF1, rawdata_ADGRF1, STAVER_ADGRF1],
target_col="ADGRF1",
titles=["Expression profiles from HPA dataset (ADGRF1)",
"Expression profiles from original data (ADGRF1)",
"Expression profiles from STAVER data (ADGRF1)"],
log_transforms=[True, False, False],
x_labels=[None, None, None],
y_labels=["Log2 (Relative expression)",
"Log2 (Relative expression)",
"Log2 (Relative expression)"],
save_path="combined_barplots_ADGRF1.pdf"
)
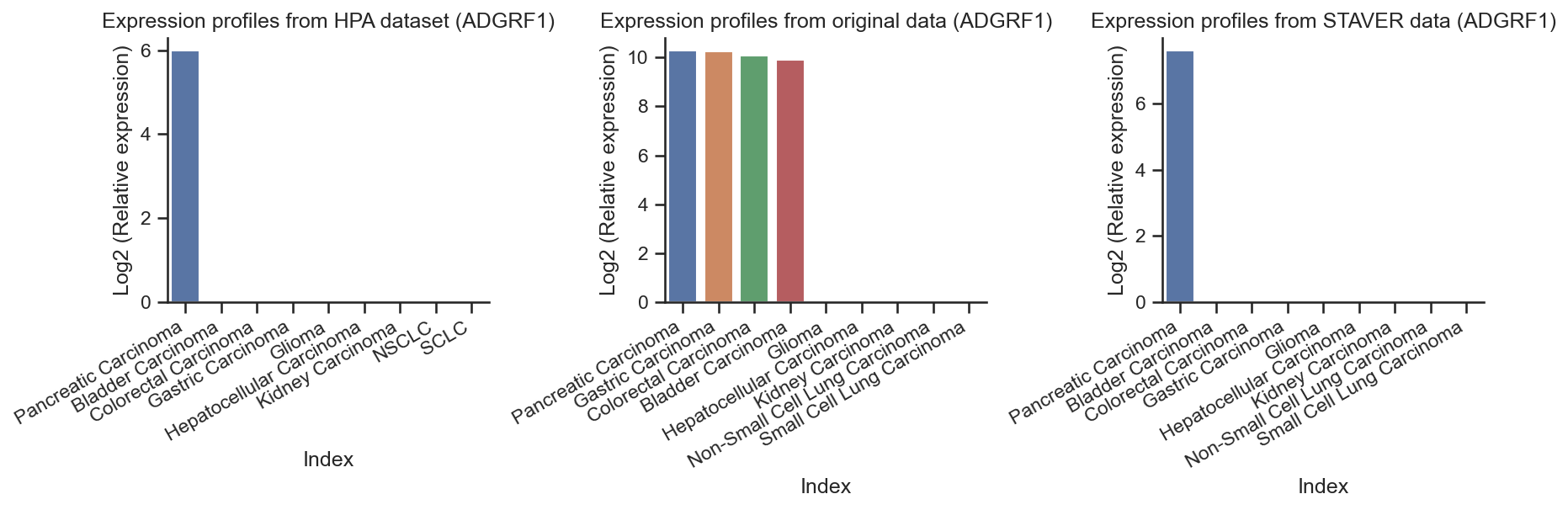
The reproducibility of the STAVER algorithm in identifying the previously reported tumor biomarkers¶
To further validate the superior reproducibility of the STAVER algorithm in identifying tumor biomarkers, a systematic literature review was conducted. According to the systematic literature review, a series of cancer biomarkers were selected to further evaluate the STAVER algorithm (Table RL1 and Table RL4). We utilized the original and STAVER-processed proteomic data to examine the expression differences of these previously reported cancer biomarkers across diverse cancer cell lines.
The results demonstrated that the STAVER algorithm more accurately identify the previously reported tumor biomarkers with high reproducibility.
[49]:
reported_biomrkers = pd.read_clipboard()
reported_biomrkers
[49]:
| Tissue Type | Cancer Type | Reported Biomarker | Reference | PMID | |
|---|---|---|---|---|---|
| 0 | Liver | Hepatocellular carcinoma | DCP | Journal of Hepatology, 2023 | PMID: 37683735 |
| 1 | Liver | Hepatocellular carcinoma | HSP70 | Journal of hepatology, 2009 | PMID: 19231003 |
| 2 | Liver | Hepatocellular carcinoma | GGT1 | Gastroenterology, 2017; BMC Cancer, 2019 | PMID: 28711626; PMID: 31455253 |
| 3 | Liver | Hepatocellular carcinoma | A1CF | Cell reports, 2019; The JCI, 2021 | PMID: 31597092; PMID: 33445170 |
| 4 | Kidney | Kidney cancer | CA9 | European urology, 2014; Cancer research, 1997 | PMID: 24821582; PMID: 9230182 |
| 5 | Kidney | Kidney cancer | CD70 | Cancer research, 2006 | PMID: 16489038 |
| 6 | Gastric | Gastric cancer | ERRB2 | Annals of oncology, 2008 | PMID: 18441328 |
| 7 | Gastric | Gastric cancer | AGR3 | In vivo, 2023 | PMID: 36593009 |
| 8 | Colorectal | Colorectal cancer | MUC2 | Gastroenterology, 2005; | PMID: 16285957 |
| 9 | Colorectal | Colorectal cancer | EPCAM | British journal of cancer, 2014 | PMID: 24786601 |
| 10 | Colorectal | Colorectal cancer | CDX2 | Annals of oncology, 2017 | PMID: 28328000 |
| 11 | Colorectal | Colorectal cancer | MUC13 | Oncogene, 2019 | PMID: 31427737 |
| 12 | Brain | Glioma | GFAP | Brain, 2007; Biosens Bioelectron, 2020 | PMID: 17998256; PMID: 33160234 |
| 13 | Brain | Glioma | NES | Cancer cell, 2020; Mol Neurobiol, 2017 | PMID: 32396858; PMID: 26768429 |
| 14 | Pancreas | Pancreatic cancer | MUC1 | Journal of gastroenterology,2003 | PMID: 14714254 |
| 15 | Pancreas | Pancreatic cancer | ANO1 | PNAS, 2019 | PMID: 31182586 |
| 16 | Lung | Non-small cell lung cancer | KRT5 | Cancer cell, 2022 | PMID: 36368318 |
| 17 | Lung | Non-small cell lung cancer | DSC3 | Journal of thoracic oncology, 2011 | PMID: 21623236 |
| 18 | Lung | Small cell lung cancer | CHGA | Cell reports, 2020 | PMID: 33086069 |
| 19 | Lung | Small cell lung cancer | NCAM1 | Cancer cell, 2022 | PMID: 36368318 |

All the results are shown in the above figure and can be reproduced with the R script boxplot_949_PanCancer.R.
The robustness and broad applicability of the STAVER algorithm for disease diagnosis and classification.¶
To validate the outstanding advantages of the STAVER algorithm for potential clinical decision-making, we comprehensively evaluated the generalization performance and prediction accuracy of classification models constructed based on the above tumor biomarkers.
To illustrate the robustness and broad applicability of the STAVER algorithm, we constructed three separate benchmark classification models based on different algorithms, including decision tree-based random forest, gradient boosting-based XGBoost, and linear regression-based logistic regression models (Materials and methods section).
As a result, compared to the original proteomic data, the classification models constructed based on STAVER-processed data exhibited increased generalization performance in distinguishing specific cancer-type cell lines.
See the PanCancer_949_cellline_models.ipynb for more details.
[ ]: